2 Apr 2018
Charlotte Maile describes the most commonly reported conditions affecting glycogen storage in domestic animals.
Charlotte Maile
Job Title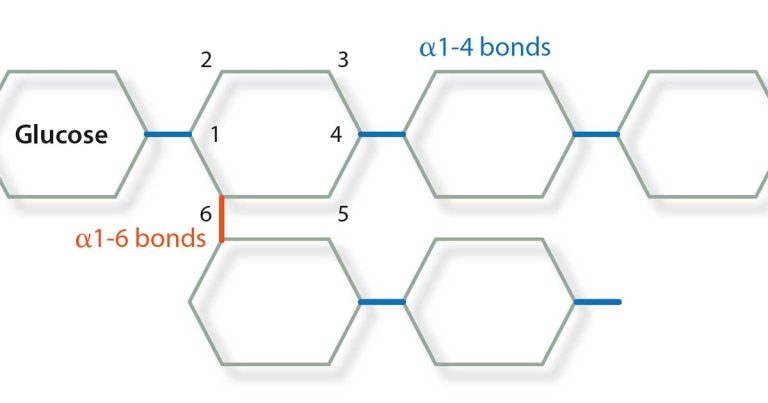
Ask any marathon runner what it feels like to “hit the wall“ and he or she will tell you it is soul destroying – it can even require you being carried over the finish line, as was seen at last year‘s London Marathon.
Glycogen – well, the lack of it – is the cause of this phenomenon and highlights just how important this complex sugar is as an energy store; defects in glycogen storage can have a catastrophic effect on metabolism and survival.
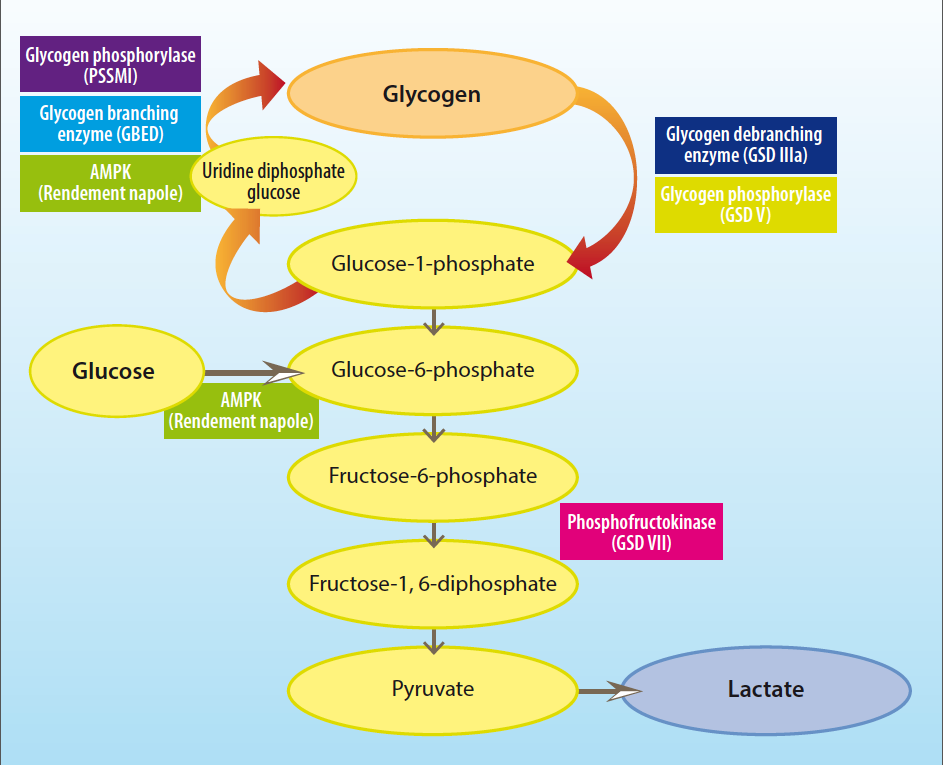
Glycogen storage diseases (GSDs) are inherited disorders due to defects in normal glycogen metabolism. A total of 14 GSDs are reported in humans and several more identified in animals (Table 1), characterised by excessive glycogen accumulation or abnormal glycogen structure. These disorders are often caused by defects in glycogenesis (the generation of glycogen) or glycogenolysis (the breakdown of glycogen; Figure 1).
A majority of GSDs are associated with excessive glycogen accumulation or abnormal glycogen structure (rather than the deficiency of it). This does, however, often result in an inability to break down the polysaccharide and, subsequently, similar symptoms, to a complete lack of the important energy store.
This article describes the GSDs most commonly reported in domestic animals.
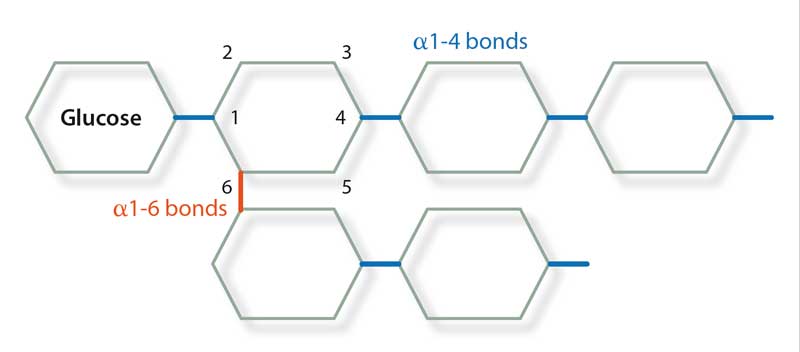
Pompe‘s disease (GSD II; numerical nomenclature of these diseases is based on human GSDs) was first identified as a human disease associated with increased glycogen storage resulting in hypertrophic cardiomyopathy and increased glycogen storage within cardiomyocytes. The disorder is caused by a deficiency of acid maltase, an enzyme that normally hydrolyses the α1-4 and α1-6 bonds in glycogen (Figure 2).
This disease has since been characterised in dogs with similar clinical signs (Mostafa, 1970) and, more recently, the associated nonsense mutation in the canine acid maltase gene was identified (Seppala et al, 2013). This disease has an autosomal recessive mode of inheritance and, therefore, can be silently passed on to offspring from two unaffected carrier parents. Genetic testing is available for this mutation and should be advised for dogs used for breeding – especially in breeds with a high prevalence of the mutation, such as Finnish Lapphunds.
GSD IIIa is reported in curly coated retrievers and is associated with a deficiency of the glycogen debranching enzyme. This results in reduced breakdown of glycogen and, subsequently, its accumulation in muscle and liver tissue. Affected dogs present with episodic exercise intolerance and collapse, and are usually detected in their first year of life with elevated liver enzymes. This disease also has an autosomal recessive mode of inheritance and, therefore, unaffected individuals can carry the mutation and pass it on to their progeny.
Tarui’s disease (GSD VII) is caused by a deficiency in the enzyme, phosphofructokinase (Tarui et al, 1965), which phosphorylates fructose-6-phosphate to form fructose-1, 6-bisphosphonate, and the committal of glucose to glycolysis. Reduced glycolysis leads to increased glycogen storage and the creation of polyglucosan bodies in muscle.
Phosphofructokinase deficiency has been reported in several canine breeds, including English springer spaniels and whippets. Dogs have similar clinical signs to humans with Tarui’s disease, including exertional myopathy and haemolytic anaemia (Smith et al, 1996).
Lafora disease is a human glycogen storage condition first described in 1911 by Gonzalo Rodriguez-Lafora – a Spanish neurologist. The disease is characterised by the presence of Lafora bodies – accumulations of poorly branched insoluble glycogen-like polymers in muscles, neurons, the liver and other tissues (Tagliabracci et al, 2008). It is caused by a mutation in a gene associated with carbohydrate metabolism – epilepsy progressive myoclonic 2b. Clinically, patients develop myoclonic epilepsy in their teenage years.
Lafora disease has also been described in dogs with myoclonic epilepsy (Holland et al, 1970) and miniature wire-haired dachshunds, beagles and basset hounds are predisposed. The molecular cause in dogs was identified prior to those in humans (Lohi et al, 2005).
Quarter horses (QHs) are relatively common sufferers of GSD with two different disorders identified in the breed – both related to enzymes involved in glycogenesis.
Polysaccharide storage myopathy (PSSM) was first described in QHs in 1992 and is the most well known GSD in horses, having since been recognised in many other breeds (Valberg et al, 1992). The prevalence of PSSM is particularly high in certain breeds, namely QHs and certain continental European draught-related breeds, and considerable variation in severity of the clinical signs exists, ranging from stiffness to severe rhabdomyolysis.
A genetic basis for PSSM was published in 2008 showing a majority of horses with PSSM carry a dominant missense mutation in the equine muscle glycogen synthase gene (GYS1), which results in a constitutively active enzyme (Maile et al, 2017). Horses with the GYS1 mutation are said to have type 1 PSSM (PSSM1) and those with the disease and without the mutation are said to have type 2 (PSSM2). Glycogen synthase catalyses the formation of the α1-4 bonds in glycogen and glycogen branching enzyme (GBE) catalyses the formation of the α1-6 bonds, therefore forming the branch points of the polysaccharide. The ratio of activity between these two enzymes is crucial to ensure normal glycogen is formed with branches every 6 to 10 residues.
Glycogen branching enzyme deficiency (GBED) is a disease reported in QH foals associated with an autosomal recessive mutation in the equine glycogen branching enzyme gene (GBE1) resulting in a premature stop codon (Valberg et al, 2001; Ward et al, 2004). Affected animals have accumulations of unbranched glycogen in multiple tissues, which is likely due to the change in GS:GBE ratio (Valberg et al, 2001). The foals showed clinical signs including transient flexural limb deformities and seizures; in some cases the foals were stillborn (Valberg et al, 2001).
Norwegian forest cats are also affected by GBED and, like the affected horses, have multisystemic accumulation of unbranched glycogen-like polysaccharide (Fyfe et al, 1992). The autosomal dominant mutation associated with GBE deficiency in cats is, however, different to the mutation found in horses. Anderson disease (GSD IV) is the human form of GBE deficiency.
McArdle disease (GSD V) was first identified in 1951 (McArdle, 1951) and is caused by a deficiency of muscle glycogen phosphorylase resulting in increased glycogen content within skeletal muscle (Mommaerts et al, 1959). Muscle glycogen phosphorylase catalyses the breakdown of glycogen: its absence or deficiency in patients with McArdle disease means they accumulate glycogen and have inadequate lactic acid production at exercise. Glycogen structure is normal in patients with McArdle disease as glycogen synthesis is not affected. A muscle glycogen phosphorylase deficiency has been documented in Charolais cattle that present with signs of severe rhabdomyolysis and exercise intolerance (Angelos et al, 1995; Tsujino et al, 1996).
In these cattle, a missense mutation in a highly conserved region of the bovine phosphorylase glycogen muscle (PYGM) gene was found (Tsujino et al, 1996) and a splice site mutation in PYGM has also been described in sheep with GSD V (Tan et al, 1997).
Increased muscle glycogen is also associated with a dominant mutation in the protein kinase adenosine monophosphate (AMP)-activated protein kinase (AMPK) γ3 subunit gene (PRKAG3) in pigs (Andersson, 2003; Milan et al, 2000). AMPK is considered a major regulator of cellular energy levels, and defects in this protein result in a multitude of consequences due to the enzyme’s wide-reaching effects. This so-called Rendement Napole (RN-) phenotype is common in Hampshire pigs and the mutation is associated with increased muscle glucose uptake and up to a 70% increase in muscle glycogen content (Andersson, 2003). The high prevalence of this mutation may be linked to the beneficial effects on meat quality in heterozygous carriers of the RN- mutation (Enfält et al, 1997; Lundstrom et al, 1998).
Several GSDs exist in animals and it is likely more are yet to be identified. Due to the heterogenous nature of the clinical signs, it is important to have GSDs on the list of differential diagnoses, especially in breeds that have a high prevalence of a known GSD.