19 Jun 2017
Albane Fauron and Karen Perry look at the physiology of disordered function associated with this condition, in the first of a three-part article.

Figure 1. Mediolateral and caudocranial views of the left stifle in addition to a photograph of the patient – a five-month-old Labrador retriever. This dog presented following a traumatic incident while out on a walk. On both the caudocranial and mediolateral views, a mineralised fragment is evident within the joint consistent with an avulsion fracture of the cranial cruciate ligament. On the mediolateral view, this is associated with evidence of pronounced synovial effusion.
Cranial cruciate ligament (CrCL) disease is the most common cause of pelvic lameness in the adult dog and typically caused by progressive ligamentous degeneration of unknown origin. Degeneration of the CrCL has been attributed to a variety of factors that may be broadly classified as genetic, conformational, environmental, immune-mediated and inflammatory; none of these, however, have definitely been proven to be causative. Further investigation into this complex and common condition is warranted to investigate whether any preventive measures may be feasible once predisposed dogs have been identified.
Cranial cruciate ligament (CrCL) disease is the most common cause of stifle-associated lameness in the adult dog (Ness et al, 1996; Johnson and Johnson, 1993) and the primary trigger of degenerative joint disease affecting the stifle (Piermattei et al, 2006).

While traumatic CrCL rupture occurs, the vast majority of cases presenting with CrCL instability are associated with progressive ligamentous degeneration of unknown cause (Moore, 1996). The initial lameness following CrCL rupture is typically non-weight-bearing, but most animals will start to use the limb within two to three weeks of the injury, if no surgical repair is performed before (Piermattei et al, 2006).
As periarticular fibrotic tissue forms, the joint becomes progressively more stable and clinical improvement will usually continue – albeit not to a normal level of function – until secondary meniscal damage and/or osteoarthritic changes develop, contributing to further functional decline (Piermattei et al, 2006).
Prevalence values of 0.56% to 2.6% have been reported for CrCL disease (Taylor-Brown et al, 2015; Witsberger et al, 2008) and the economic impact of the management of this condition has been estimated to be US$1.32 billion (£1.03 billion) annually in the US alone (Wilke et al, 2005). However, despite the cost and morbidity associated with CrCL disease, our limited understanding of the complex and likely multifactorial aetiology of non-traumatic CrCL rupture has considerably limited development of preventive measures.
CrCL rupture can result from acute traumatic rupture secondary to excessive strain or progressive degeneration of unknown cause (Kowaleski, 2012).
Traumatic acute rupture of the CrCL typically results from hyperextension and/or excessive internal tibial rotation (Kowaleski, 2012) – similar to the situation in the majority of people with anterior cruciate ligament (ACL) rupture. In both scenarios, the tibial excursion results in CrCL overload, causing rupture. Hyperextension probably occurs most commonly by stepping into a hole or depression at a fast gait while excessive tibial rotation occurs when the dog rapidly turns towards the limb with the foot firmly planted (Piermattei et al, 2006).
Figure 1 shows radiographs and an image of a young Labrador retriever that presented with a traumatic CrCL avulsion after getting its foot stuck in a hole while running. However, trauma accounts for a minority of cases in dogs, with only 20% of cases presenting with CrCL-associated instability being attributable to a traumatic event (Moore, 1996).
More commonly, dog CrCL deficiency is associated with a progressive degeneration of unknown origin. Histologic examination of ruptured ligaments shows alteration to the cell population and extracellular matrix (ECM) structure of the CrCL.
In a comparative histological study, CrCL rupture was associated with significant loss of ligament fibroblasts from the core region (the major axial tissue component) of the ligament (Hayashi et al, 2003). Moreover, this decrease in cell density was further characterised by chondroid metaplasia of the surviving fibroblasts, resulting in fibroblasts similar in appearance to the articular chondrocytes often seen in late-stage osteoarthritic cartilage.
ECM changes included extensive disruption to the architecture of the collagen, with decreased birefringence and elongation of crimping in the remaining collagen fibrils, suggesting progressive mechanical overload as a cause of failure. Additionally, Vasseur et al found the aforementioned degenerative changes to be more pronounced and occur at an earlier age in dogs weighing more than 15kg when compared to dogs weighing less than 15kg, which is consistent with the observation cruciate disease tends to occur at a younger age in large breed dogs (Vasseur et al, 1985; Whitehair, 1993; Duval, 1998).
The underlying cause of this degeneration remains unclear. While the regulatory mechanism of differentiation from ligament cells to chondrocytes has not been identified, Hayashi et al suggested it may be part of a protective response to hypoxia or oxidative stress, as has been suggested for osteoarthritic cartilage (Grimshaw and Mason, 2001; Schipani et al, 2001; Hayashi et al, 2003). In a more recent study, Ichinoe et al (2015) suggested production of cartilage matrix and fibrocyte metaplasia might be a response to some external force, such as microinjury. The same authors also reported increased collagen III expression in ruptured CrCL fibres, which is the first step in injury healing; this further supports the theory that microinjury could potentially promote the degenerative changes observed.
However, to date – and despite extensive research – the exact cause and pathogenesis of CrCL deficiency remains unclear. This is largely due to the complex and likely multifactorial origin of the disease process. Over the years, progressive degeneration of the CrCL has been attributed to a variety of additional factors that may be broadly classified as genetic, conformational, environmental, immune-mediated and inflammatory; none of these, however, has definitely proved to be causative.
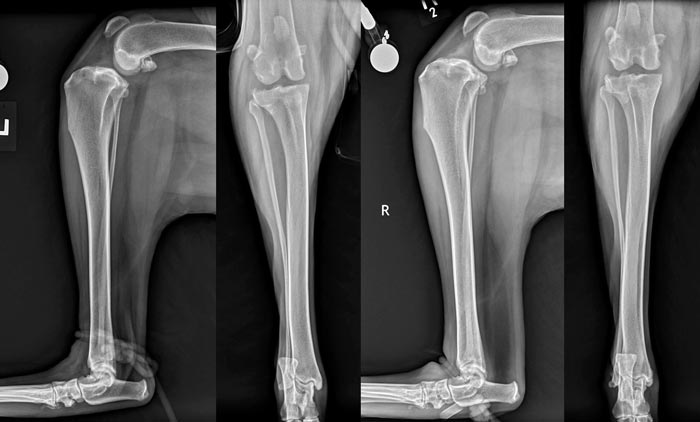
Overrepresentation of certain breeds in cases of non-traumatic CrCL deficiency, along with the high prevalence of bilateral cases, have been the rationale behind exploring the existence of a genetic component to CrCL disease.
The heritability index (HI) is commonly used as an estimate of heritability. It ranges from zero to one; an HI of zero indicates none of the variability of the trait of interest among individuals is the result of genetic factors, while an HI of one indicates all of the variability of that trait among the group of individuals is the result of genetic factors. Another commonly used term when talking about genetics is the penetrance that indicates the proportion of individuals carrying a mutated gene that will go on to exhibit clinical signs. The authors of one study focusing on a population of 574 Newfoundlands proposed a recessive mode of inheritance for CrCL deficiency, with 51% penetrance and a heritability estimate of 0.27 (Wilke et al, 2006).
Figure 2 shows bilateral stifle radiographs for a two-year-old Newfoundland that presented with bilateral CrCL disease. A heritability of 0.28 was also identified in a cohort of boxers (Nielen et al, 2003). While both studies support a genetic component to the disease process, environmental factors likely remain a major influence in the incidence of CrCL disease. In an additional study focusing on the Newfoundland breed, Wilke et al (2009) found several microsatellite markers associated with CrCL rupture on chromosomes 3, 5 and 13.
A subsequent genome-wide association study conducted in Newfoundland pedigree dogs with and without CrCL disease failed to find any significant associations in either chromosome 5 or 13, but did identify single nucleotide polymorphisms on chromosomes 1, 3 and 33, which could potentially be associated with CrCL deficiency (Baird et al, 2014).
Further fine mapping and functional analyses of these regions may narrow the list of CrCL deficiency genes in future (Baird et al, 2014). At this stage, whether genetics exert a direct influence on the structural properties of the CrCL or control conformation factors that predispose to CrCL deficiency is unclear (Griffon, 2010).
Conformational factors that have been implicated with CrCL deficiency include an upright pelvic limb stance, genu varum (“bow-legged stance”), narrowing of the distal intercondylar notch, increased tibial plateau angle and conformation of the tibial tuberosity.
The role of the CrCL is to prevent cranial displacement of the tibia relative to the femur, prevent stifle hyperextension and limit internal tibial rotation (Arnoczky and Marshall, 1977). Hence, it is possible the presence of a “straight” (hyperextended) stifle joint exacerbates the underlying degenerative process and predisposes to early CrCL rupture (Johnson and Johnson, 1993). This assumption was, however, initially based on clinical observations and has not been well substantiated, largely because of its inconsistent presence in dogs with CrCL deficiency and difficulties in obtaining objective measurements (Griffon, 2010).
Genum varum is associated with femoral varus of varying severity, as well as internal rotation of the tibia, and has been associated with other conditions, such as medial patellar luxation (MPL; Kowaleski et al, 2012; Harasen, 2006). Concomitance of CrCL pathology and MPL is well recognised, with 13% to 25% of dogs presenting for MPL found to have parallel CrCL rupture (Gibson et al, 2006; Campbell et al, 2010). It has been suggested dogs with MPL have an increased risk of developing CrCL disease due to malalignment of the extensor mechanism and internal rotation of the proximal tibia, which, in turn, puts continuous tension on the CrCL, predisposing it to deterioration (Gibson et al, 2006; Harasen, 2002).
In addition, MPL can be associated with degenerative joint disease (DJD) that produces an enzymatic environment that can lead to degradation of the CrCL (Gibson et al, 2006; Roy et al, 1992). It is also speculated dogs with severe MPL may have atrophy of the supporting structures of the stifle joint as a result of severe lameness. Atrophy of structures, such as the vastus lateralis muscle or the lateral retinaculum – combined with a lack of patellar ligament stability – may contribute to greater joint instability and greater tension on the CrCL (Campbell et al, 2010).
Several studies support presence of a narrow intercondylar notch (ICN) in dogs with CrCL deficiency, potentially constantly or intermittently impinging on the CrCL. Intercondylar stenosis and its association with anterior cruciate ligament (ACL) rupture in people has been well documented (Shelbourne et al, 1998; LaPrade and Burnett, 1994; Stein et al, 2010).
Aiken et al (1995) were the first to report similar findings in the dog and found cranial cruciate deficient stifles had significantly smaller ICN compared to normal stifles. Comerford et al (2006) showed ICN dimensions to be greater in stifles harvested from greyhounds (a breed rarely affected by CrCL rupture) than in Labrador retrievers and golden retrievers – two breeds known to be predisposed to early CrCL degeneration. They concluded impingement of the CrCL by the ICN in high risk breeds may result in collagen remodeling and the aforementioned subsequent histopathological degenerative changes eventually causing reduced structural integrity of the ligament and its rupture.
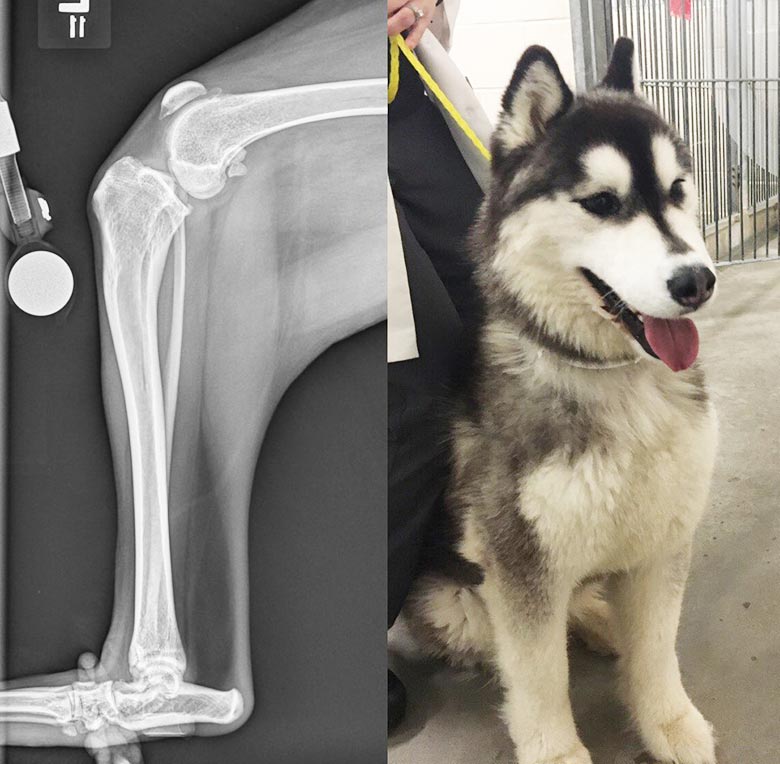
It has been suggested excessive cranial tibial thrust due to a steep tibial plateau angle (TPA) could contribute to early failure of the CrCL (Slocum and Devine, 1983; Korvick et al, 1994).
Figure 3 is a radiograph and an image of a young dog that presented with CrCL disease associated with an excessive TPA. In a clinical study measuring and comparing TPAs of dogs with CrCL injuries and dogs without, Morris and Lipowitz (2001) found the mean TPA in the CrCL rupture group was 5.7° greater than the mean TPA of the normal group, suggesting a possible causative effect of increased TPA on CrCL deficiency. However, additional investigations have failed to substantiate these findings.
Reif and Probst (2003) found no significant difference between Labrador retrievers with and without CrCL deficiency, while Wilke et al (2002) compared standing and traditional TPAs in Labrador retrievers (high-risk breed) and greyhounds (low-risk breed) and found, though Labrador retrievers had a mean TPA greater than those of greyhounds, the steepest TPA was found in clinically unaffected Labrador retrievers.
The discrepancy between studies likely results from variations in methods and interbreed variation, as well as differences in selection criteria for normal dogs. This illustrates the difficulty in identifying the role of a single factor in the aetiology of a likely multifactorial disease. Moreover, accurately comparing the TPA of dogs with and without CrCL rupture can be challenging as it relies on correct and consistent radiographic technique, and is subject to interobserver and intraobserver variation (Caylor et al, 2001).
Additionally, in the absence of a definitive primary cause for CrCL disease, identifying a control group of normal individuals remains difficult (Reif and Probst, 2003).
Ultimately, the true effect of TPA on CrCL stresses in vivo remains unknown as other factors can influence the amount of stress sustained by the CrCL in addition to the TPA (Colborne et al, 2005). While TPA has never clearly been identified as a single causative factor in CrCL rupture, it may still contribute to early ligament rupture in the presence of other concomitant predisposing factors.
A retrospective study performed on 219 stifles found tibial tuberosity (TT) width to be significantly smaller in CrCL deficient stifles when compared to healthy stifles, suggesting TT conformation may also be a risk factor for CrCL rupture (Inaeun et al, 2009).
The authors hypothesised a small size TT relative to the rest of the structures around the stifle joint could theoretically reduce the angle of insertion of the patellar ligament and lead to increased cranial tibial thrust. This, in turn, may predispose to more rapid CrCL deterioration, leading to rupture in a younger dog population.
Studies have shown female and neutered dogs to be at increased risk of CrCL rupture (Witsberger et al, 2008; Whitehair et al, 1993; Duval et al, 1999). In one, neutered female dogs had 2.1 times the odds of being diagnosed with CrCL compared with entire females (Taylor-Brown et al, 2015). A possible association may exist between the increased prevalence of obesity seen among neutered females and the high prevalence of CrCL disease in this population (Edney and Smith, 1986).
Obese dogs are almost four times as likely to suffer CrCL disease as those of “normal weight” (Adams et al, 2011). However, a study of CrCL disease in a UK population of dogs found no significant difference in the body condition score (BCS) of neutered dogs compared to their entire counterparts, suggesting a lack of association between neutering status and obesity (Adams et al, 2011). The underlying reason for the increased incidence of CrCL disease observed in neutered animals might require further investigation.
Regardless of neutering status, Taylor-Brown et al (2015) found increased bodyweight in dogs to be associated with increased risk of CrCL rupture. As body size is intrinsic to the breed, the study avoided confounding breed and body size effects by comparing between bodyweight tertiles within breeds and found dogs in the heaviest third of bodyweight to be 3.4 times more likely to suffer CrCL disease.
A higher BCS may result in abnormal tension on articular and periarticular structures, and yield a propensity for ligament rupture. In addition to mechanical overload of the stifle joint, obesity could also contribute to CrCL rupture via the release of potentially proinflammatory mediators. Indeed, it is hypothesised excess white adipose tissue in obese individuals leads to a persistent state of chronic inflammation, hereby contributing to the aetiopathogenesis of connective tissue pathology via the release of key proinflammatory adipokines (Trayhurn et al, 2006; Eisele et al, 2005).
However, a possible role for systemic adipose tissue in the development of CrCL rupture has not been examined and requires further investigation (Comerford et al, 2011).
The CrCL is mainly composed of collagen type I and the demonstration of anticollagen type I and type II antibodies in both the sera and synovial fluid of dogs with CrCL disease led some authors to speculate immunologic reactivity may play a role in CrCL disease, with the higher incidence of these antibodies in the synovial fluid suggestive of local antibody production (Niebauer and Menzel, 1982; Niebauer et al, 1987).
However, a later study comparing synovial fluid samples from clinically normal dogs and dogs with diseased stifles suffering from either CrCL or DJD secondary to other arthropathies found increases in autoantibodies to collagen in synovial fluid to not be specific for the type of joint disorder (Rooster et al, 2000).
A more recent prospective study, in which anticollagen type I antibody synovial titres were sequentially measured over a 12 to 18-month period, did not provide evidence anticollagen antibodies initiated CrCL damage in dogs (de Bruin et al, 2007). Measurements were performed on 13 dogs that initially presented with unilateral CrCL rupture and 2 clinically normal dogs. Six of 13 dogs sustained contralateral CrCL rupture during the study; however, not all dogs with all high antibody titres developed contralateral rupture.
Additionally, among the six dogs that did, two had very low or undetectable antibody titres in the contralateral stifle before subsequent ligament rupture. Nevertheless, it is possible anticollagen antibodies perpetuate chronic joint inflammation in some dogs with cruciate deterioration, even if collagen is not the primary arthritogenic agent (Doom et al, 2008).
In addition to immune-mediated mechanisms, considerable interest has also been seen in the role of joint inflammation in the pathogenesis of CrCL disease.
The reason why the secondary DJD typically seen in the cruciate deficient stifle continues to develop despite restoration of joint stability is still not understood, but has led to the suggestion biomechanical factors in addition to biomechanical stresses are responsible for these changes (Cameron et al, 1997; Rayward et al, 2004). Biomechanical factors thought to induce and perpetuate joint inflammation are proinflammatory cytokines.
While human studies in patients with ACL injury susceptible to cartilage loss and DJD have shown an imbalance between pro and anti-inflammatory cytokines, to date limited information exists on the role of cytokines and growth factors in canine degenerative joint disease (Fernihough et al, 2003). In an attempt to further understand how DJD may contribute to progressive CrCL degradation and eventual rupture, Muir et al (2005a) looked at the pattern of collagenolytic gene expression in the ruptured CrCL and synovial fluid and found increased expression of matrix degrading enzymes.
Release of collagenolytic proteases into the synovial fluid can significantly degrade the structural properties of the CCL and predispose it to rupture (Muir et al, 2002; Muir et al, 2005a,b). However, it remains to be determined if changes in these proteolytic enzymes initiate cruciate disease in dogs or if they are the result of disease progression (Doom et al, 2008).
CrCL deficiency is a complex and multifactorial disease yet to be fully understood. Genetics may have a direct influence on the structural properties of the CrCL or influence other factors contributing to CrCL deficiency, such as abnormal stifle conformation. Abnormal biomechanics may, in turn, lead to microinjury to the CrCL, leading to compositional and structural changes within the ligament ECM.
The synovitis and DJD typically seen in these cases may partially initiate, contribute and/or propagate an inflammatory cascade contributing to CrCL degradation and eventual rupture. This field of research is wide open and, as further investigations deepen our understanding of this multifaceted condition, future preventive measures and treatment options may include specific and sensitive detection of predisposed dogs and immune-modulating therapies in dogs with early evidence of CrCL disease.
The next part of this article will examine the epidemiology of CrCL disease, detail the signalment of affected dogs and present how a diagnosis of a partial or full CrCL tear is reached. The third part will present treatment options; indications for surgical stabilisation will be considered in addition to the prognoses associated with the different treatment options.